Cluster of Hungarian Medical Manufacturers and Service Providers (http://mediklaszter.eu/en)
Mirbest Central European Gastroinnovation Cluster (https://www.mirbest.hu/en/
Hungarian Biotechnology Association (http://hungarianbiotech.org/?
Cluster of Hungarian Medical Manufacturers and Service Providers (http://mediklaszter.eu/en)
Mirbest Central European Gastroinnovation Cluster (https://www.mirbest.hu/en/
Hungarian Biotechnology Association (http://hungarianbiotech.org/?
Next-generation sequencing techniques allow us to generate reads from a microbial environment in order to analyze the microbial community. However, assembling of a set of mixed reads from different species to form contigs is a bottleneck of metagenomic research. Although there are many assemblers for assembling reads from a single genome, there are few assemblers for assembling reads in metagenomic data without reference genome sequences. Moreover, the performances of these assemblers on metagenomic data are far from satisfactory, because of the existence of common regions in the genomes of subspecies and species, which make the assembly problem much more complicated. To solve this problem we use the latest, best benchmarked algorithms for assembling reads in metagenomic data, which contain multiple genomes from different species.
 |
The main problem addressed by MEGAN is to compute a “species profile” by assigning the reads from a metagenomics sequencing experiment to appropriate taxa in the NCBI taxonomy. At present, this program implements the following naive approach to this problem:
1. Compare a given set of DNA reads to a database of known sequences, such as NCBI-NR or NCBI-NT, using a sequence comparison tool such as BLAST.
2. Process this data to determine all hits of taxa by reads.
3. For each read r, let H be the set of all taxa that r hits.
4. Find the lowest node v in the NCBI taxonomy that encompasses the set of hit taxa H and assign the read r to the taxon represented by v.
We call this the naive LCA-assignment algorithm (LCA = “lowest common ancestor”). In this approach, every read is assigned to some taxon. If the read aligns very specifically only to a single taxon, then it is assigned to that taxon. The less specifically a read hits taxa, the higher up in the taxonomy it is placed. Reads that hit ubiquitously may even be assigned to the root node of the NCBI taxonomy.
If a read has significant matches to two different taxa a and b, where a is an ancestor of b in the NCBI taxonomy, then the match to the ancestor a is discarded and only the more specific match to b is used.
The program provides a threshold for the bit disjointScore of hits. Any hit that falls below the threshold is discarded. Secondly, a threshold can be set to discard any hit whose disjointScore falls below a given percentage of the best hit. Finally, a third threshold is used to report only taxa that are hit by a minimal number of reads or minimal percent of all assigned reads. By default, the program requires at least 0:1% of all assigned reads to hit a taxon, before that taxon is deemed present. All reads that are initially assigned to a taxon that is not deemed present are pushed up the taxonomy until a node is reached that has enough reads. This is set using the Min Support Percent or Min Support item.
Taxa in the NCBI taxonomy can be excluded from the analysis. For example, taxa listed under root - unclassified sequences - metagenomes may give rise to matches that force the algorithm to place reads on the root node of the taxonomy. This feature is controlled by Preferences!Taxon Disabling menu. At present, the set of disabled taxa is saved as a program property and not as part of a MEGAN document.
Note that the LCA-assignment algorithm is already used on a smaller scale when parsing individual blast matches. This is because an entry in a reference database may have more than one taxon associated with it. For example, in the NCBI-NR database, an entry may be associated with up to 1000 different taxa. This implies, in particular, that a read that may be assigned to a high level node (even the root node), even though it only has one significant hit, if the corresponding reference sequence is associated with a number of very different species.
Note that the list of disabled taxa is also taken into consideration when parsing a BLAST file. Any taxa that are disabled are ignored when attempting to determine the taxon associated with a match, unless all recognized names are disabled, in which case the disabled names are used.
The weighted LCA algorithm is identical to the weighted LCA algorithm used in Metascope. It operates as follows: In a first round of analysis, each reference sequence is given a weight. This is the number of reads that align to the given reference and that have the property that all the significant alignments for the read are to the same species as the reference sequence (but can also be to a strain or sub-species below the species node). In a second round of analysis, each read is placed on the node that is above 75% of the total weight of all references for which the read has a significant alignment.
The Weighted LCA algorithm will assign reads more specifically than the naive LCA algorithm. Because it performs two rounds of read and match analysis, it takes twice as long as the naive algorithm.
The weighted LCA algorithm is identical to the weighted LCA algorithm used in Metascope. It operates as follows: In a first round of analysis, each reference sequence is given a weight. This is the number of reads that align to the given reference and that have the property that all the significant alignments for the read are to the same species as the reference sequence (but can also be to a strain or sub-species below the species node). In a second round of analysis, each read is placed on the node that is above 75% of the total weight of all references for which the read has a significant alignment.
The Weighted LCA algorithm will assign reads more specifically than the naive LCA algorithm. Because it performs two rounds of read and match analysis, it takes twice as long as the naive algorithm.
Allows to interactively explore and analyze, both taxonomically and functionally, large-scale microbiome sequencing data. MEGAN is a comprehensive microbiome analysis toolbox for metagenome, metatranscriptome, amplicon and from other sources data. User can perform taxonomic, functional or comparative analysis, map reads to reference sequences, reference-based multiple alignments and reference-guided assembly and integrate their own classifications.
However, the main application of the program is to parse and analyze the result of an alignment of a set of reads against one or more reference databases, typically using BLASTN, BLASTX or BLASTP or similar tools to compare against NCBI-NT, NCBI-NR or genome specific databases. The result of a such an analysis is an estimation of the taxonomical content (“species profile”) of the sample from which the reads were collected. The program uses a number of different algorithms to “place” reads into the taxonomy by assigning each read to a taxon at some level in the NCBI hierarchy, based on their hits to known sequences, as recorded in the alignment file.
Alternatively, MEGAN can also parse files generated by the RDP website or the Silva website. Moreover, MEGAN can parse files in SAM format. MEGAN provides functional analysis using a number of different classification systems. The InterPro2GO viewer is based on the Gene Ontology metagenomic goslim and the InterPro database. The eggNOG viewer is based on the “eggNOG” database of orthologous groups and functional annotation.
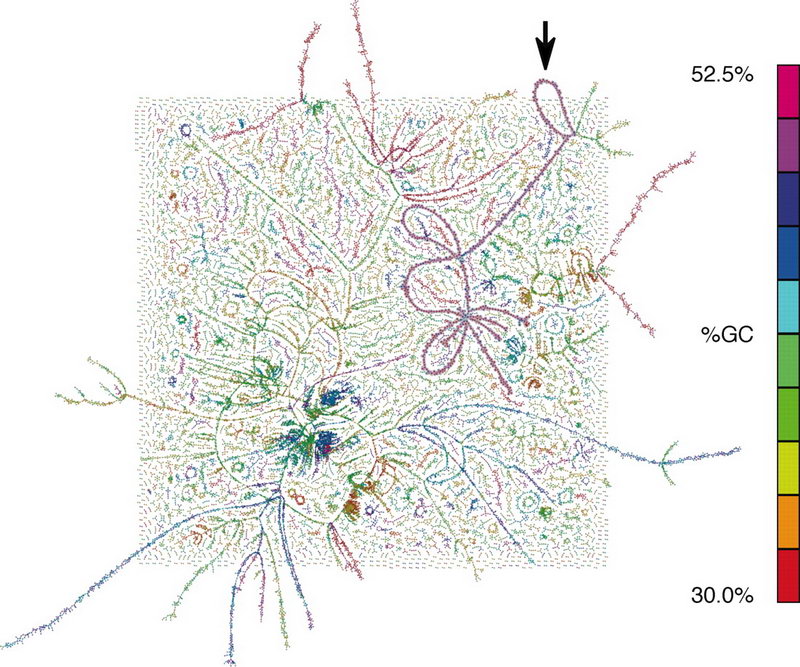
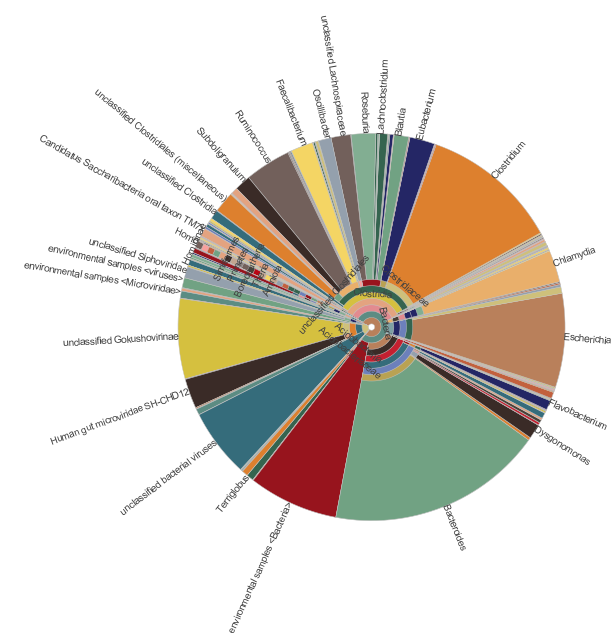
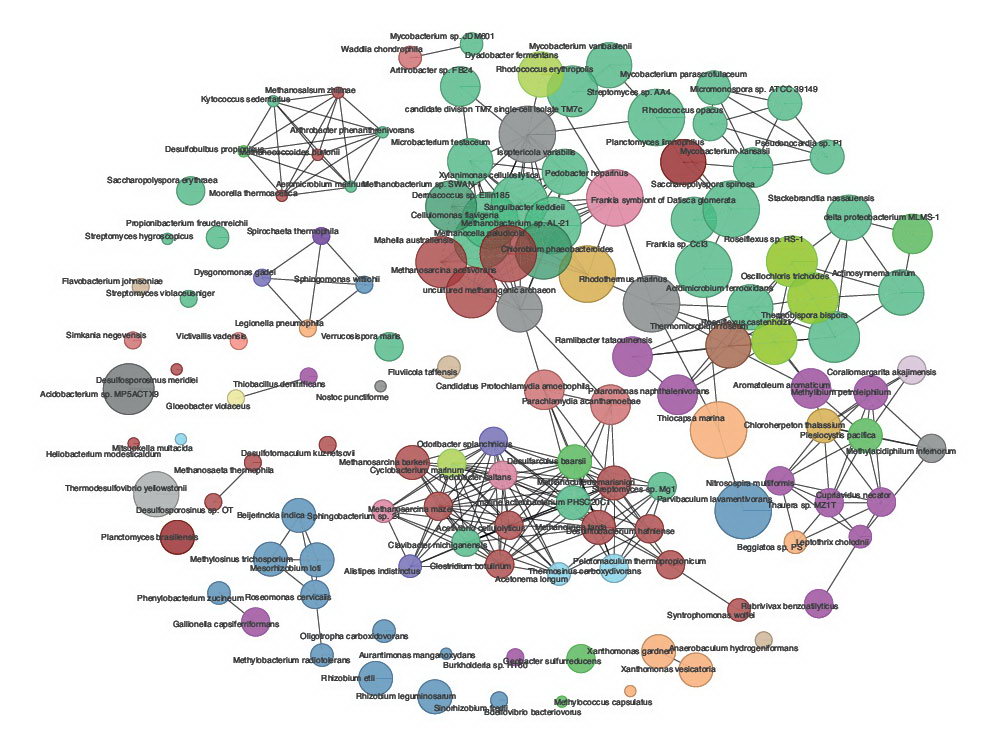
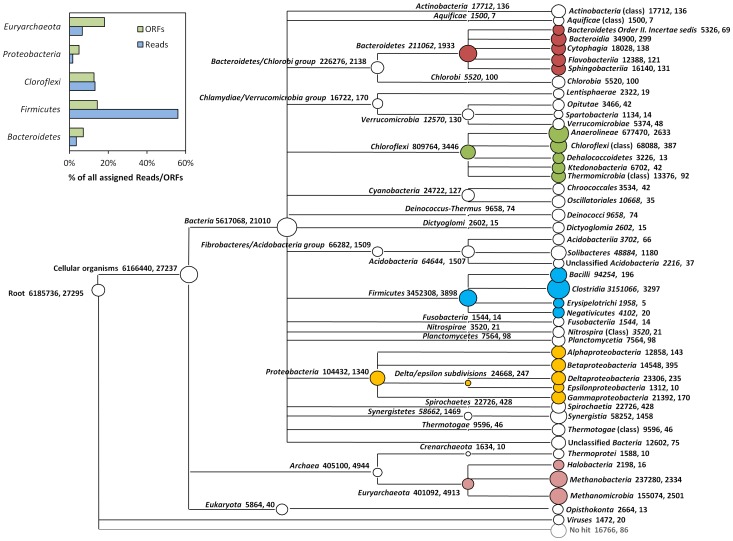
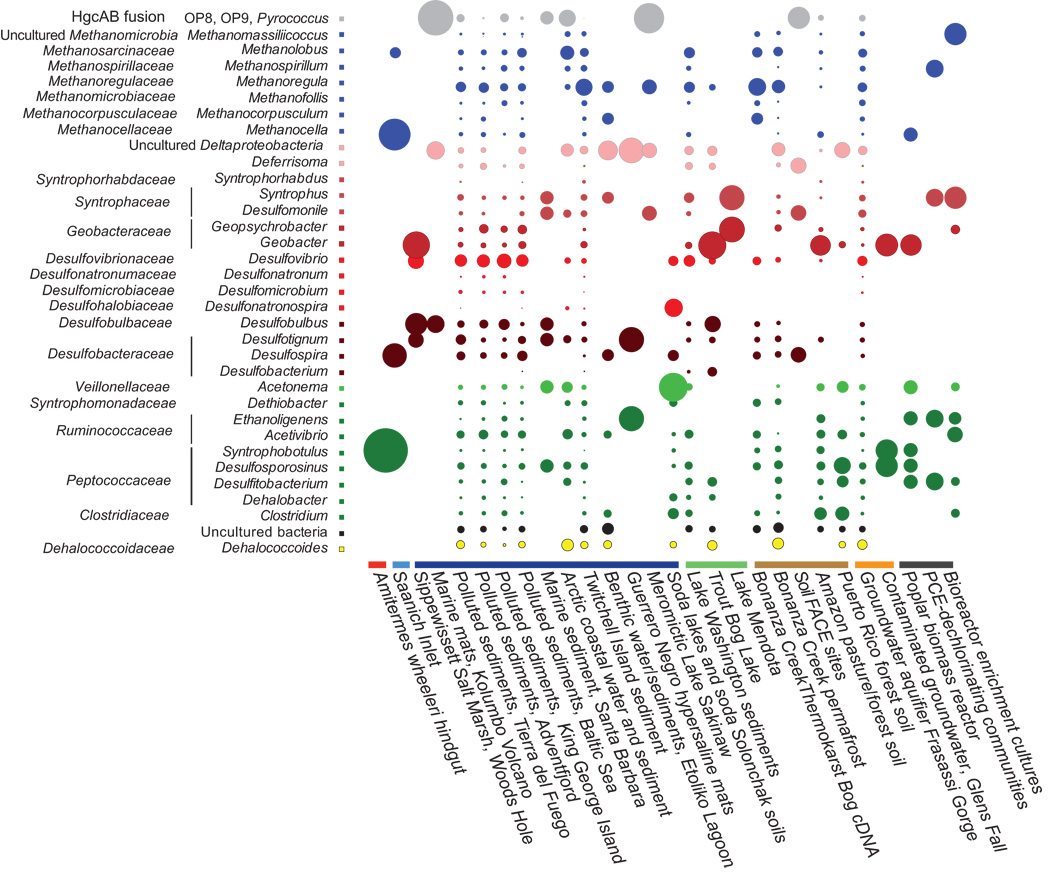
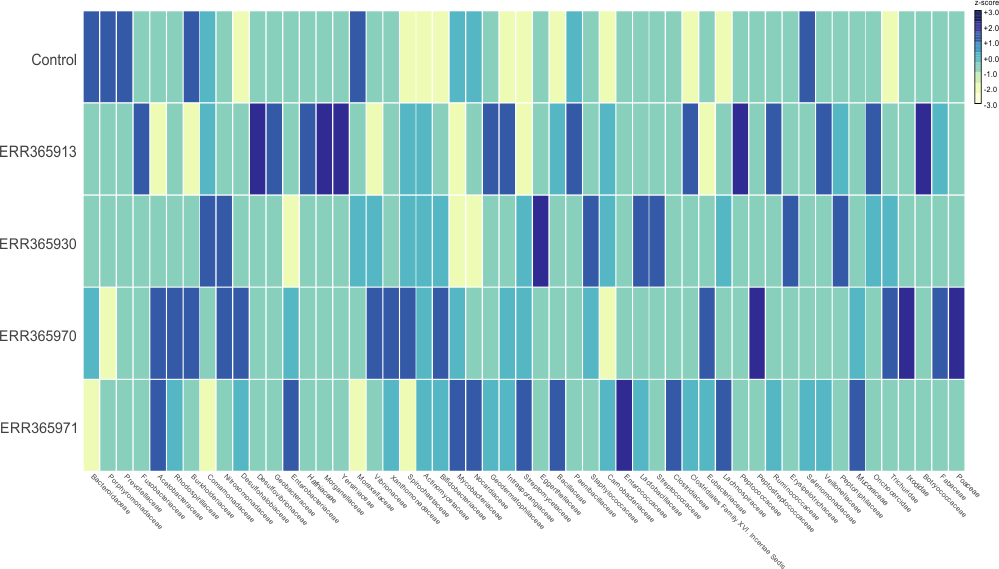
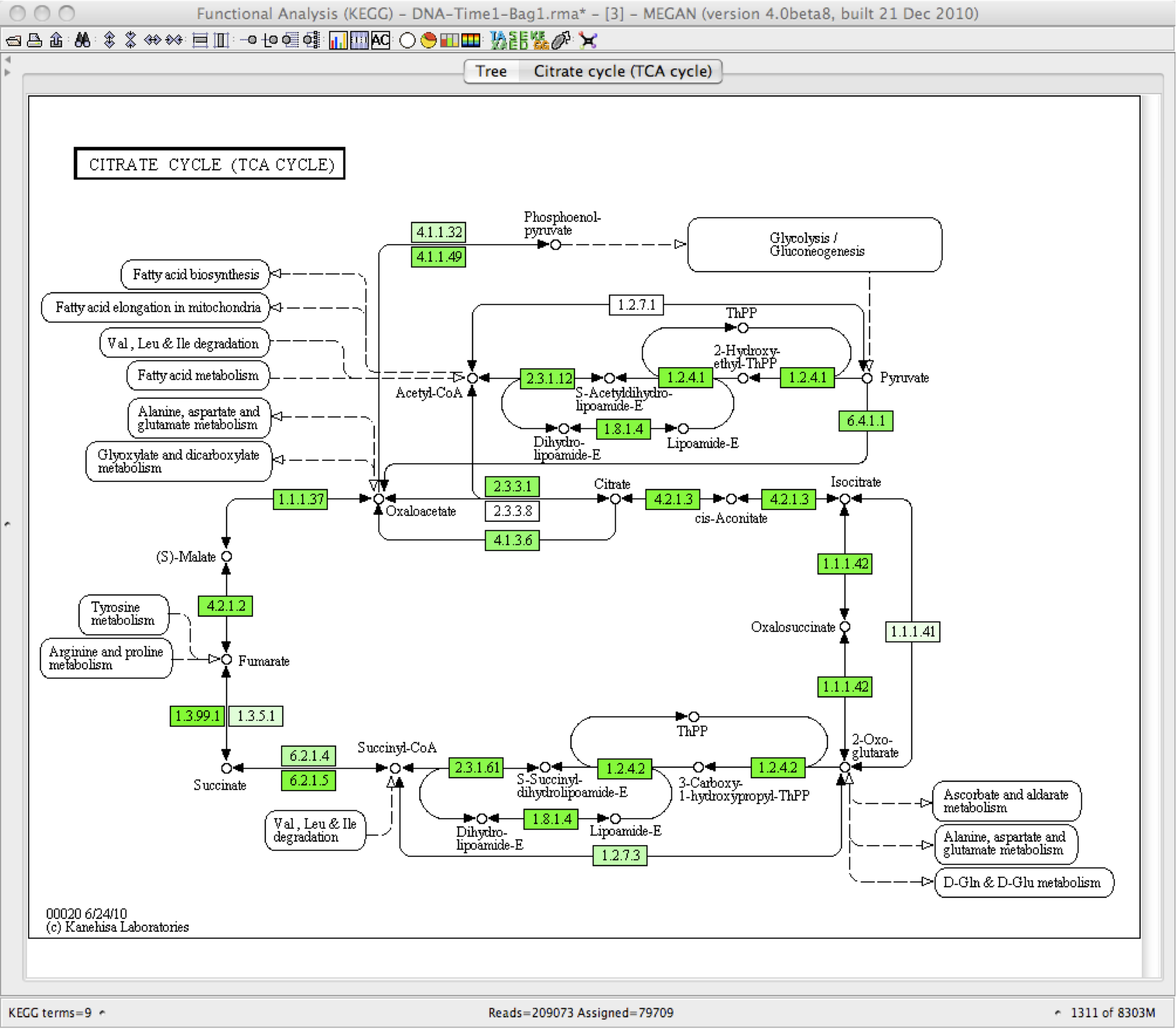
Some example of the various visualisations generated by MEGAN:
 |
 |
 |
 |
 |
 |